- About us
- Research
- Students & Teaching
- Seminars & Events
- Directories
- Booking Rooms & Equipment
- עברית
Home » Dr. Dina Schneidman, Prof. Michal Linial and Dr. Nir Kalisman study the "social network" of the Corona virus
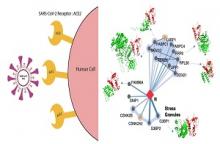
All viruses make extensive use of the host capacity for completing the infectious cycle from the initial attachment to the host receptor until the release of 100s infectious viruses. To identify the Achilles heel of SARS-CoV-2 that can be utilized as a drug target, it is crucial to obtain detailed structural models of the interactome of the viral protein cross-talk with the host. Recently, large-scale interactome covering all 29 viral proteins was reported with hundreds of candidate pairs that might be useful therapeutic targets. The challenge is to identify the most therapeutically attractive virus-host pairs and exclude those that are likely to fail in drug design effort.
Identification of druggable targets requires structural characterization of the virus-host protein complexes. Even with a high structural coverage of the viral proteome, experimental characterization of the hundreds of the virus-host complexes is beyond reach of current technologies. We propose to bridge the gap through integration of structural modeling, mass spectrometry, molecular dynamics, and genomic variability.
In a study that is currently being conducted in collaboration by the research groups of Dina Schneidman, Nir Kalisman and Michal Linial, we are attacking the task from complementary directions. First, we use structural modeling to characterize virus-host interactions. As viral proteins often resemble native human interactors, our modeling uses known protein-protein interactions (PPIs) as templates to explore such molecular mimicry mechanisms. Second, we rely on Molecular Dynamics (MD) simulations to reveal the temporal dimension and the effect of genomic variations. We can analyze the short-term evolution of the COVID-19 virus, which like other RNA-viruses undergoes constant genetic changes that reflect its high mutation rate. Third, we will provide a set of experimentally validated interactions by utilizing a novel approach based on in situ cross-linking coupled to mass spectrometry (XL-MS). The method expects to reveal novel interactions and validate previously reported pairs. In situ XL-MS offers a great advantage of reporting on protein interactions as they occur in the intact cell, overcoming the noisy data due to non-specific interactions. We will employ in situ XL-MS coupled with affinity purification and formaldehyde cross-linker that efficiently penetrates cell membranes. Importantly, the cross-linking data will enable us to significantly improve the accuracy of the structural models.
We have recently applied structural modeling and MD simulations to characterize the SARS-CoV-2 spike receptor-binding domain and its interaction with the human ACE2 receptor. By comparison to other coronaviruses of natural and designed origins, we found that the ACE2 binding interface mutates faster than the rest of the virus, thus it is not a preferred drug target and any future drug and vaccine will need to cope with the evolution. The results were published recently in the Viruses journal.